My TreeBASE R package is essentially functional now. Here’s a quick tutorial on the kinds of things it can do. Grab the treebase package here, install and load the library into R.
TreeBASE provides two APIs to query the database, one which searches by the metadata associated with different publications (called OAI-PMH), and another which queries the phylogenies directly (called Phylo-ws). They have somewhat redundant functions, but for our purposes the second one returns the actual data, while the first returns metadata. A few examples will best illustrate how this all works. We start with some queries of the metadata directly without downloading any trees.
Trees can be downloaded with search_treebase(), using a variety of search conditions provided by Phylo-ws API.
Metadata can be searched by date using search_metadata. Just Download all metadata for finer queries.
Metadata for a study can be grabbed using the study id.
A few examples will better illustrate how this all works.
Metadata queries
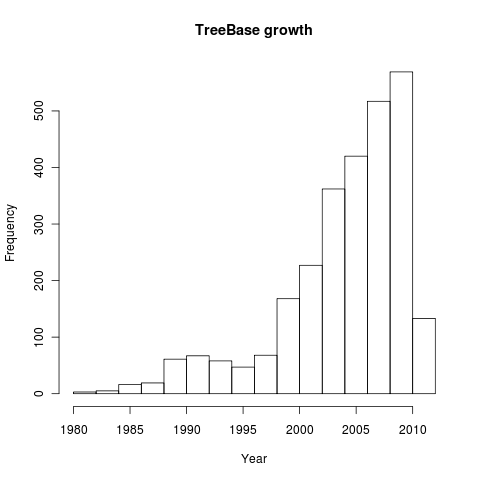
How has TreeBASE grown since it’s inception? Let’s grab the metadata for all entries and histogram by publication year:
all <- search_metadata("", by="all")
dates <- sapply(all, function(x) as.numeric(x$date))
hist(dates, main="TreeBase growth", xlab="Year")
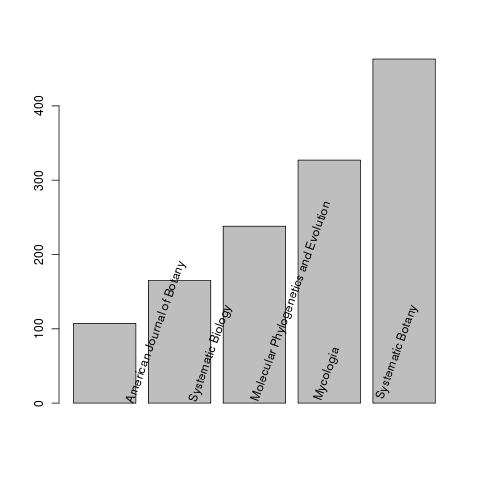
What journals have submitted the most studies to treebase?
journals <- sapply(all, function(x) x$publisher)
J <- tail(sort(table(as.factor(unlist(journals)))),5)
b <- barplot(as.numeric(J))
text(b, names(J), srt=70, pos=4, xpd=T)
How many have been submitted from Nature? Science?
nature <- sapply(all, function(x) length(grep("Nature", x$publisher))>0)
science <- sapply(all, function(x) length(grep("^Science$", x$publisher))>0)
sum(nature)
> 11
sum(science)
> 8
Which studies were those? Can I have those trees please?
s <- get_study_id( all[nature] )
nature_trees <- sapply(s, function(x) search_treebase(x, "id.study"))
Other details associated with the study are certainly also available. Since we downloaded all metadata we have this stored already. Any tree downloaded stores the TreeBASE study id in $S.id, ((and the unique tree id in $Tr.id)) which we can use to look up the metadata again.
plot(nature_trees[[1]][[1]]) #plot first tree in the first study in the set
all[nature][1] # Pull metadata from complete list
# Or look up again using the study id:
metadata(nature_trees[[1]][[1]]$S.id)
Importing Phylogenies
We can query for phylogenies directly on an array of search criteria, such as study authors, taxa included, number of taxa, number of characters in the trait matrix, etc, as illustrated by the following examples. A few queries that aren’t built into the API (i.e. does the tree have branch lengths) are accomplished by filtering after downloading the tree, which is slower.
## We'll often use max_trees in the example so that they run quickly,
# notice the quotes for species.
dolphins <- search_treebase('"Delphinus"', by="taxon", max_trees=5)
## can do exact matches
humans <- search_treebase('"Homo sapiens"', by="taxon", exact_match=TRUE, max_trees=10)
# all trees with 5 taxa
five <- search_treebase(5, by="ntax", max_trees = 10)
# These are different, a tree id isn't a Study id. we report both
studies <- search_treebase("2377", by="id.study")
tree <- search_treebase("2377", by="id.tree")
c("TreeID" = tree$Tr.id, "StudyID" = tree$S.id)
# Only results wiht branch lengths
# Has to grab all the trees first, then toss out ones without branch_lengths
Near <- search_treebase("Near", "author", branch_lengths=TRUE)
length(Near)
These queries can be combined with metadata searches
#### Metadata examples ###
# Use the OAI-PMH api to check out the metadata from the study in which tree is published:
metadata(Near[[1]]$S.id)
# or manualy give a sudy id
metadata("2377")Combining metadata and phylogeny queries
Metadata queries can optionally return only those studies added to TreeBASE before or after a given date:
# Use that to get all trees "published" after 2010
# publication date is only a year
post2010 <- sapply(dates, function(x) 2010 < as.numeric(x))
s <- get_study_id( all[post2010] )
out <- lapply(s, function(x) search_treebase(x, "id.study"))
# Grab the trees entered since 2011: (some studies will have multiple trees)
#can compare dates with as.Date("2001-01-01", "%y-%m-%d) < as.Date ...
m <- search_metadata("2011-05-05", by="from")
s <- get_study_id(m)
out <- lapply(s, function(x) search_treebase(x, "id.study"))A simple meta-analysis
Of course, this capacity is most powerful not to merely get some summary statistics of the database, but repeat analyses of given studies or perform meta-analyses. Most comparative phylogenetics methods require ultrametric trees.
We can assemble a simple pipeline to perform the meta-analysis across all existing studies of whether phylogenies tend to fit a pure-birth or a birth-death model more frequently:
As a proof-of-principle, we can create a pipeline that will estimate chronograms for all trees containing branchlengths in treebase.
timetree <- function(tree){
check.na <- try(sum(is.na(tree$edge.length))>0)
if(is(check.na, "try-error") | check.na)
NULL
else
try( chronoMPL(multi2di(tree)) )
}
drop_errors <- function(trees){
## apply to a list of trees created with timetree to drop errors
tt <- tt[!sapply(trees, is.null)]
tt <- tt[!sapply(tt, function(x) is(x, "try-error"))]
print(paste("dropped", length(trees)-length(tt), "trees"))
tt
}
require(laser)
pick_branching_model <- function(tree){
m1 <- try(pureBirth(branching.times(tree)))
m2 <- try(bd(branching.times(tree)))
as.logical(try(m2$aic < m1$aic))
}
# Return all treebase trees that have branch lengths
# This has to download every tree in treebase, so not superfast...
all <- search_treebase("Consensus", "type.tree", branch_lengths=TRUE)
tt <- drop_errors(sapply(all, timetree))
is_yule <- sapply(tt, pick_branching_model)
table(is_yule)Replicating individual studies
Replicating individual studies is a bit more challenging, mostly do to the quality of available data. For instance, here is a nice recent study (Rowe et. al. 2011) that has nicely time calibrated chronograms (from BEAST, Figure 4) and species trees (using BEST, Figure 3), but it seems only theMr. Bayes tree in Figure 2 is given in TreeBASE.
Further, replicating the study would require other data than the phylogenetic tree. In the future we might hope that this data would appear on Dryad. Dryad’s web-based search frustratingly does not seem to allow a simple query by doi or treebase id, and a query by title returns a long list of things that aren’t this paper.
The Mendeley API (through my RMendeley package) does a bit better at pulling out some metadata, but not much. Querying by the doi we can’t find the paper, but the pmid works:
require(RMendeley)
details("21239388", "pmid")
Some Dryad papershave phylogenies, and the data deposition includes the BEAST xml files necessary to reproduce the phylogenies, but not, it seems, the phylogenies themselves. It would be great to have some good examples of papers with data up on both TreeBASE and Dryad. Further, a pipeline that could regenerate the trees from the alignments might be interesting.
RaXML for inferring branch lengths: A quick solution for trees that have only topologies would be to infer branch lengths conditional on the topology from the character matrix. TreeBASE does not include this with the nexus file containing the tree, and would require a separate query to pull the character matrix. This is handled in the read.nexus.data command.
References
- Rowe K, Aplin K, Baverstock P and Moritz C (2011). “Recent And Rapid Speciation With Limited Morphological Disparity in The Genus Rattus.” Systematic Biology, 60. ISSN 1063-5157, https://dx.doi.org/10.1093/sysbio/syq092.